HOME > English > Hirohisa HIRAI
Hirohisa HIRAI

Molecular cytogenetics and Chromosome evolution in primates
Professor
Hirohisa HIRAI
Primate Research Institute, Kyoto University, Inuyama, Aichi 484-8506, Japan
Tel: +81-568-63-0528, Fax: +81-0568-63-0085,
E-mail:hirai.hirohisa.7w@kyoto-u.ac.jp
Academic Experience
1979-1992 Research Associate, Kumamoto University, Medical School, Japan
1987-1989 Post-doctoral Fellow and Research Assistant Professor, State University of New York at Buffalo, U.S.A.
1992-1999 Instructor, Primate Research Institute, Kyoto University, Japan
2000-2005 Associate Professor, Primate Research Institute, Kyoto University, Japan
2005-present Professor, Primate Research Institute, Kyoto University, Japan
Research interest
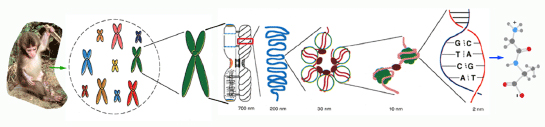
Molecular cytogenetics and Chromosome evolution in primates
Chromosomes of Primates
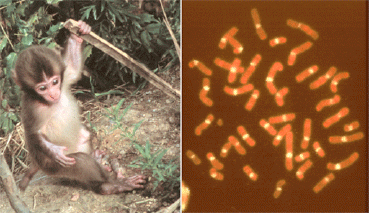 Japanese macaque(2n = 42, centromeres detected by alpha satelitte DNA)
Japanese macaque(2n = 42, centromeres detected by alpha satelitte DNA)

Aye-aye(2n = 30, conventional stain)

Blue Sykes monkey(2n = 70, C-band)
Current main researchs
(1) Night monkey hybrids exhibit de novo genomic and karyotypic alterations: the first such case in primates
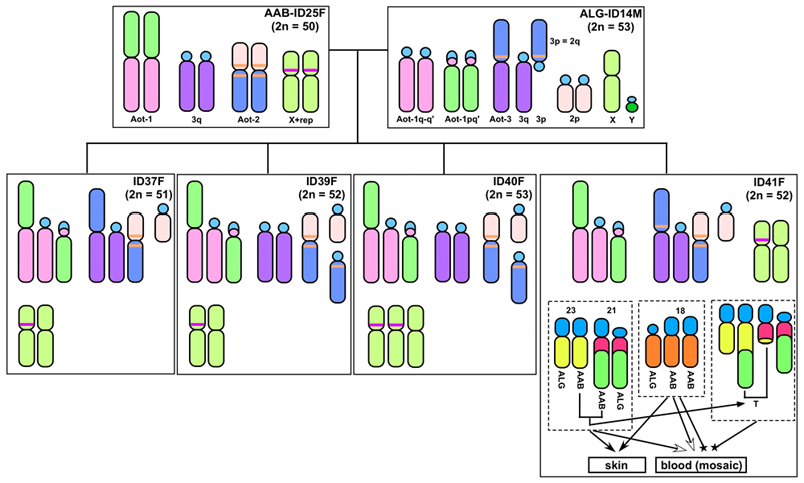
Using molecular chromosomal analyses, we discovered night monkey hybrids produced in captivity from matings between a female Aotus azarae boliviensis (2n = 50) and a male A. lemurinus griseimembra (2n = 53). The parents produced seven offspring in total, including one male and six females - a pattern consistent with Haldane's rule. Chromosomal studies were conducted on four of the hybrid offspring. Two of them showed relatively 'simple' mixture karyotypes, including different chromosome numbers (2n = 51, 52), which were formed because of a heteromorphic autosome pair in the father (n = 26, 27). The other two hybrid monkeys exhibited de novo genomic and karyotypic alterations. Detailed analysis of the alterations revealed that one individual carried a mixture karyotype of the two parental species and an X chromosome trisomy (53, XXX). The second individual displayed trisomy of chromosome 18 (52,XX,+18) and a reciprocal translocation between autosomes 21 and 23 (52,XX,+18,t(21;23)). Interestingly, the second monkey exhibited mosaicism among blood cells (mos52,XX,+18[87]/52,XX,+18,t(21;23)[85]) but only a single karyotype (52,XX,+18) in skin fibroblast cells. The X- and 18-trisomies were derived from a doubling of the mother's chromosomes in early embryonic cell division, and the reciprocal translocation likely developed in the bone marrow of the offspring, considering that it was observed only in blood cells. Such occurrence of trisomies in hybrid individuals is a unique finding in placental mammals.
(2) Hypervariability of Nucleolus Organizer Regions in Bengal Slow Lorises, Nycticebus bengalensis (Primates, Lorisidae)
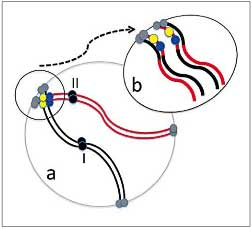
Slow lorises are a cryptic species complex, and thus genetic markers are needed to identify distinct evolutionary lineages or species. We examined the nucleolus organizer regions (NORs) of Bengal slow lorises (Nycticebus bengalensis) using FISH with 18S rDNA (rDNA-FISH) and silver nitrate staining (Ag-NOR stain). Ten individuals of the putatively single species N. bengalensis showed higher variability in localization than 3 other congeners, though their overall karyotypes were similar. The rDNA-FISH analysis detected a total of 18 loci, in contrast to previous studies of other slow loris species that revealed far fewer (6-10) loci. Eight of the 18 loci detected in the present analysis were found to be semi-stable localizations at 4 different chromosomes, while 10 were found to be unstable localizations at 5 other chromosomes. The semi-stable locations showed occasional presence/absence of variations for rDNA-FISH, and unstable locations were polymorphic among individuals, contributing to the higher variability of NORs in this taxon. We hypothesize that the larger numbers of rDNA loci found in N. bengalensis were introduced by genomic dispersion through ectopic recombination in association with terminal regions including rDNA. Such differences are potentially very powerful chromosomal markers to be used in species identification and conservation. (Cytogenetic and Genome Research)